Software & Resources

Amber
Amber is a suite of biomolecular simulation programs. It began in the late 1970's, and is maintained by an active development community; see our history page and our contributors page for more information. The term "Amber" refers to two things. First, it is a set of molecular mechanical force fields for the simulation of biomolecules; these force fields are in the public domain, and are used in a variety of simulation programs. Second, it is a package of molecular simulation programs which includes source code and demos.
Amber is distributed in two parts: AmberTools24 and Amber24. See the Download Amber link for information on how to download the code. Amber is developed in an active collaboration of David Case at Rutgers University, Tom Cheatham at the University of Utah, Ray Luo at UC Irvine, Ken Merz at Michigan State University, Maria Nagan at Stony Brook, Dan Roe at NIH, Adrian Roitberg at the University of Florida, Carlos Simmerling at Stony Brook, Junmei Wang at the University of Pittsburgh, Darrin York at Rutgers University, and many others. Amber was originally developed under the leadership of Peter Kollm


ANM
ANM (Anisotropic network model) is a simple tool for predicting the collective motions of molecular systems introduced using Elastic Network Models (ENM) and normal mode analysis. The biomolecular system is represented as a network, or graph, the nodes of which are the residues and the springs are their interactions. The model helps elucidate the intrinsic dynamics of proteins and their complexes and make inferences on functional mechanisms.
BalestraWeb
Efficient identification protein-drug interactions, including side effects and repurposable drugs is an important challenge that is likely to accelerate drug discovery and development. BalestraWeb addresses this need by learning a probabilistic latent factor model of drug-target interactions and enabling users to predict the interactions of: any known drug (listed in , DrugBank) against all targets (by providing only a drug identifier); any target protein against all known, approved drugs (by providing only a target identifier); any drug-target pair (by providing both a drug and a target identifier). BalestraWeb is built using GraphLab's collaborative filtering toolkit implementation, DrugBank data, NumPy's matrix and vector operation libraries, Flask web application framework and Python programming language, with interactive network visualizations made using Data-Driven Documents JavaScript library and Cytoscape for the static visualization of the entire drug-target interaction network.
QuartataWeb
Data on protein-drug and protein-chemical interactions are rapidly accumulating in databases such as DrugBank and STITCH. Recent studies highlight the promiscuity of both proteins and small molecules: many drugs have side effects; and many proteins bind chemicals other than those known, opening the way to design repurposable drugs, or polypharmacological treatments. There is a need for efficiently identifying such effects The QuartataWeb server is designed to address this need by learning probabilistic latent factor models for protein-drug/chemical interactions and enabling users to efficiently mine known interactions and predict new ones. QuartataWeb links targets to KEGG pathways and Gene Ontology (GO), thus completing the bridge between drugs/chemicals and pathways.



DynOmics
The DynOmics ENM server computes biomolecular systems dynamics for user-uploaded structural coordinates or PDB identifiers, by integrating two widely used elastic network models (ENMs) – the Gaussian Network Model (GNM) and the Anisotropic Network Model (ANM). Unique features include the consideration of environment, the prediction of potential functional sites and reconstruction of all-atom conformers from deformed coarse-grained structures. For more information see Theory and Tutorial.
CellBlender
CellBlender is a Blender addon for creation, simulation, visualization, and analysis of realistic 3D Cell Models. CellBlender leverages the full-featured 3D content creation capabilities of Blender to support a rich environment for the creation of simulation-ready, biophysically realistic models of the microscopic structure and biochemical function of cells.
CellBlender is fully functional with MCell and partially functional with SBML. We invite the computational cell biology community to contribute to the project, adding features and support for their favorite simulation environments.
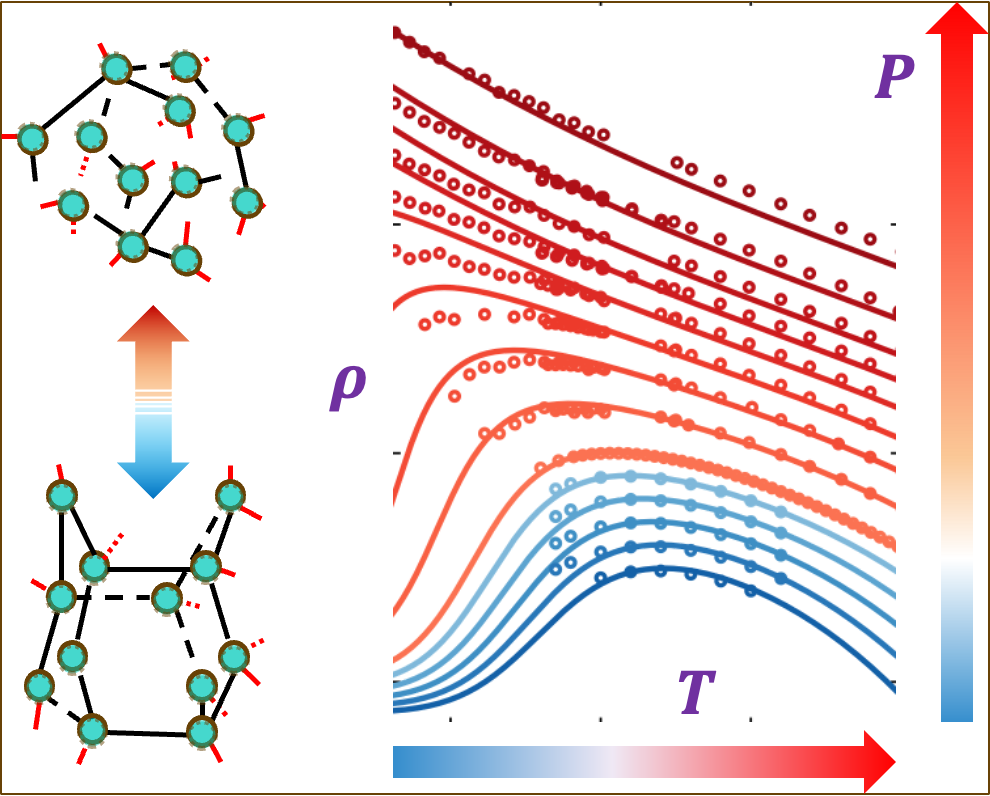
Cage Water
Cage Water is a fast analytical model of water based on three bonding states: van der Waals, pairwise hydrogen bonding, and cooperative caging hydrogen bonds. Despite its simplicity and low cost, it matches the accuracy of advanced water models such as TIP4P/2005 and MB-pol while providing an interpretable explanation for water’s anomalies, including the liquid–liquid phase transition (LLPT)
repository link: https://github.com/verlaksh/CageWater_StatMech_Water.git
iGNM
Gaussian network model (GNM) is a simple yet powerful model for investigating the dynamics of proteins and their complexes. iGNM is a user-friendly interface and database, the updated version of which, iGNM 2.0, covers a large portion of structures currently available in the Protein Data Bank (PDB). Advanced search and visualization capabilities, both 2D and 3D, permit users to retrieve information on inter-residue and inter-domain cross-correlations, cooperative modes of motion, the location of hinge sites and energy localization spots.
NEUROBLOX
Neuroblox.jl is designed for computational neuroscience and psychiatry applications. Our tools range from control circuit system identification to brain circuit simulations bridging scales from spiking neurons to fMRI-derived circuits, parameter-fitting models to neuroimaging data, interactions between the brain and other physiological systems, experimental optimization, and scientific machine learning.
MCell
MCell is a Monte Carlo reaction-diffusion simulator for modeling computational microphysiology in arbitrarily complex 3D spatial geometries.
As a modeling tool, MCell creates realistic simulations of cellular signaling in the complex 3-D subcellular microenvironment in and around living cells -- what we call cellular microphysiology. At such small subcellular scales the familiar macroscopic concept of concentration is not useful and stochastic behavior dominates. MCell uses highly optimized Monte Carlo algorithms to track the stochastic behavior of discrete molecules in space and time as they diffuse and interact with other discrete effector molecules (e.g. ion channels, enzymes, transporters) heterogeneously distributed within the 3-D geometry of the subcellular environment.
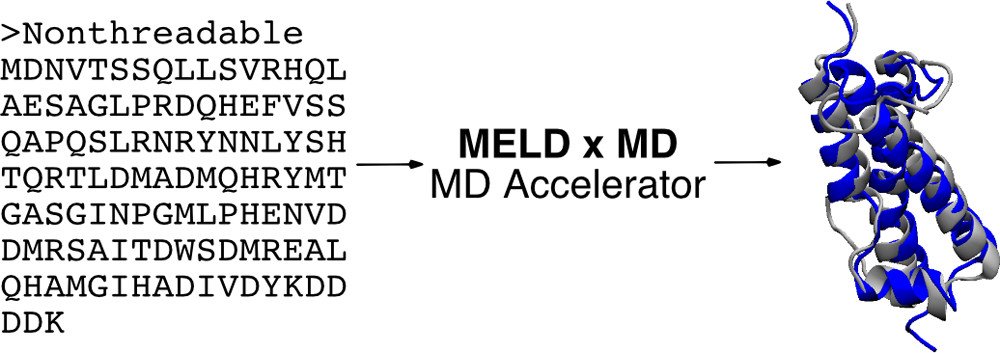
MELD x MD
In essence, modeling employing limited data (MELD x MD) provides a customizable way of running replica exchange molecular dynamics (REMD). Along the replica ladder, the temperature, hamiltonian or both can be changed. The MELD x MD code is written in Python and works closely with the OpenMM for MD simulations to provide a highly-parallelizable implementation that can run efficiently on GPU resources.
HPSandbox
HPSandbox is a package of Python modules and example scripts for experimenting with the two-dimensional HP lattice model of Dill and Chan. It is ideally used as a teaching tool, or as a way to quickly prototype 2D lattice simulation ideas with easy-to-use extensible code -- a "sandbox" if you will.
ProDy
ProDy is a free and open-source Python package for protein structural dynamics and sequence evolution analysis. It is a flexible and responsive API suitable for interactive usage and application development. NMWiz, a VMD plugin GUI, accompanies ProDy for enabling comparative visual analysis of experimental and theoretical data. With ProDy, you can perform principal component analysis of heterogeneous X-ray structures, NMR models, and MD snapshots. Protein dynamics can be modeled using normal mode analysis of anisotropic network model . Powerful and customizable atom selections allow for contact identification and comparing multiple structures/chains. ProDy contains several modules developed for sequence analysis (Evol), cryo-EM structure analysis (CryoDy), essential site scanning analysis (ESSA), druggability simulations (DRUGUI), pharmacophore modeling (Pharmmaker), and perturbation response scanning (PRS).
Rhapsody
The biological effects of human missense variants have been studied for decades but predicting their effects in clinical molecular diagnostics remains challenging. Rhapsody is a machine learning-based tool developed for predicting the effect (neutral or deleterious) of human missense variants based on sequence, structure and dynamics of proteins. The tool drew attention to the significance of structural dynamics in determining the effect of point mutations.
WESTPA
WESTPA (the Weighted Ensemble Simulation Toolkit with Parallelization and Analysis) is an open-source software package that provides a high-performance framework for carrying out extended-timescale simulations of rare events with rigorous kinetics using the weighted ensemble algorithm of Huber and Kim (1996). The software also includes options for further enhancing the sampling efficiency through reassignment of weights according to either equilibrium or nonequilibrium steady state, and a plugin for using a weighted ensemble-based string method. The software is designed to interface with any stochastic simulation engine, including but not limited to molecular dynamics (e.g. AMBER, GROMACS, and NAMD), Monte Carlo codes, BioNetGen, and MCell.