
AI in Biophysics
Is the mutation in your genome neutral or potentially disease-causing?
A new AI tool, Rhapsody-2, introduced by the Bahar lab answers this question with > 90% accuracy
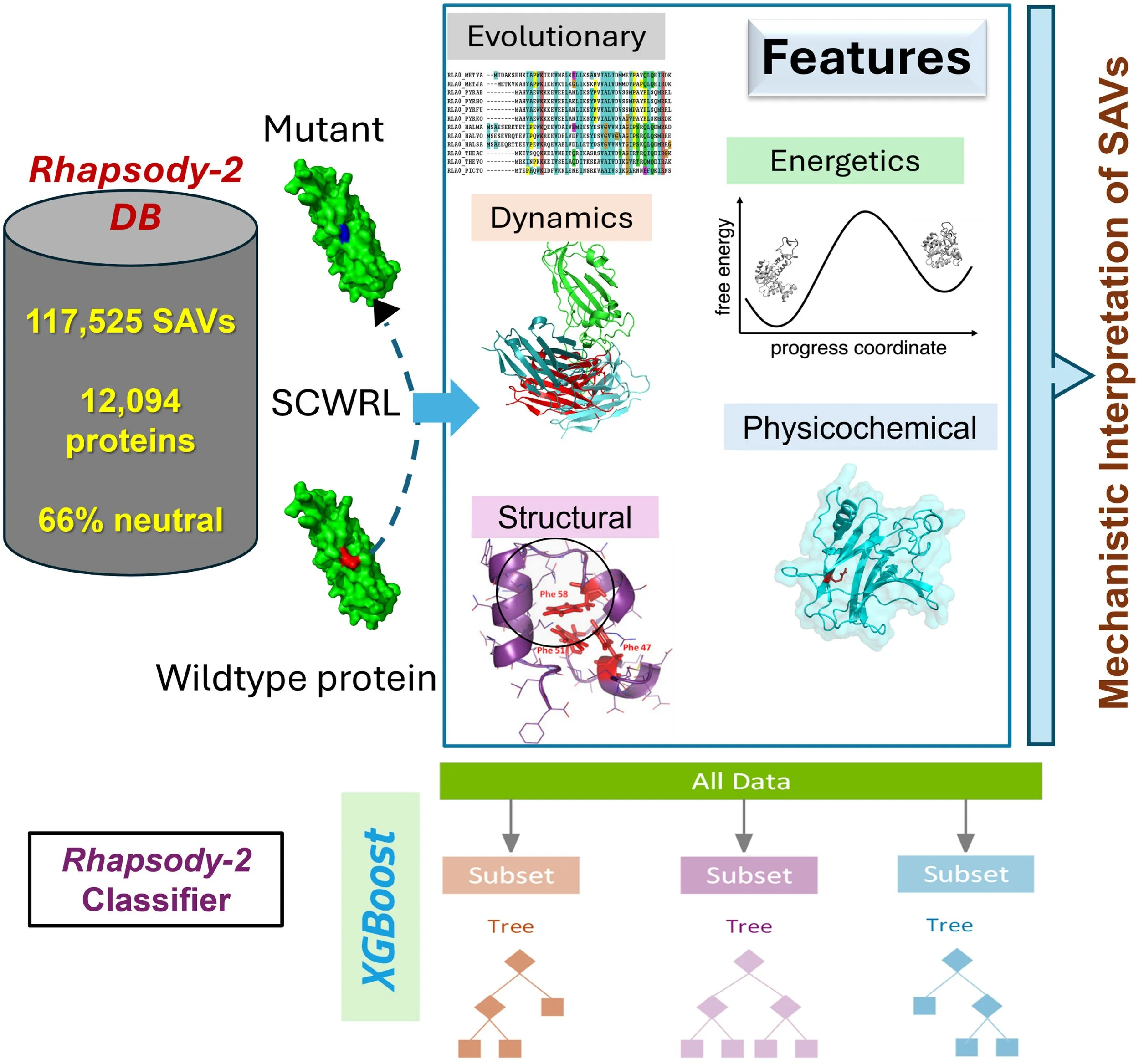
Rhapsody-2 is a cutting-edge machine learning framework that leverages AlphaFold2-predicted protein structures to enable proteome-wide prediction of pathogenic single amino acid variants (SAVs). Trained on more than 117,000 ClinVar-annotated variants across over 12,000 human proteins, it integrates evolutionary, structural, energetic, and dynamic features into a unified model. This comprehensive design delivers exceptional accuracy, achieving a cross-validated AUROC of 0.94 under stringent protein-stratified validation, and sets a new benchmark for variant classification across independent datasets, including AlphaMissense ClinVar, DDD, and cancer hotspot variants.
What distinguishes Rhapsody-2 is its ability to move beyond prediction toward mechanistic explanation. While evolutionary descriptors dominate in predictive strength, dynamics-based features—capturing fluctuations in motion, constraints in soft modes, and disruptions of allosteric communication—provide transparent insight into how mutations alter protein function. By uniting predictive power with mechanistic clarity, Rhapsody-2 demonstrates the synergy of artificial intelligence and structural biophysics, advancing precision medicine by making variant pathogenicity both highly accurate and biologically interpretable.
Ref: A Banerjee, AT Bogetti, I Bahar (2025). Accurate identification and mechanistic evaluation of pathogenic missense variants with Rhapsody-2 , Proc Natl Acad Sci USA 122 (18), e2418100122
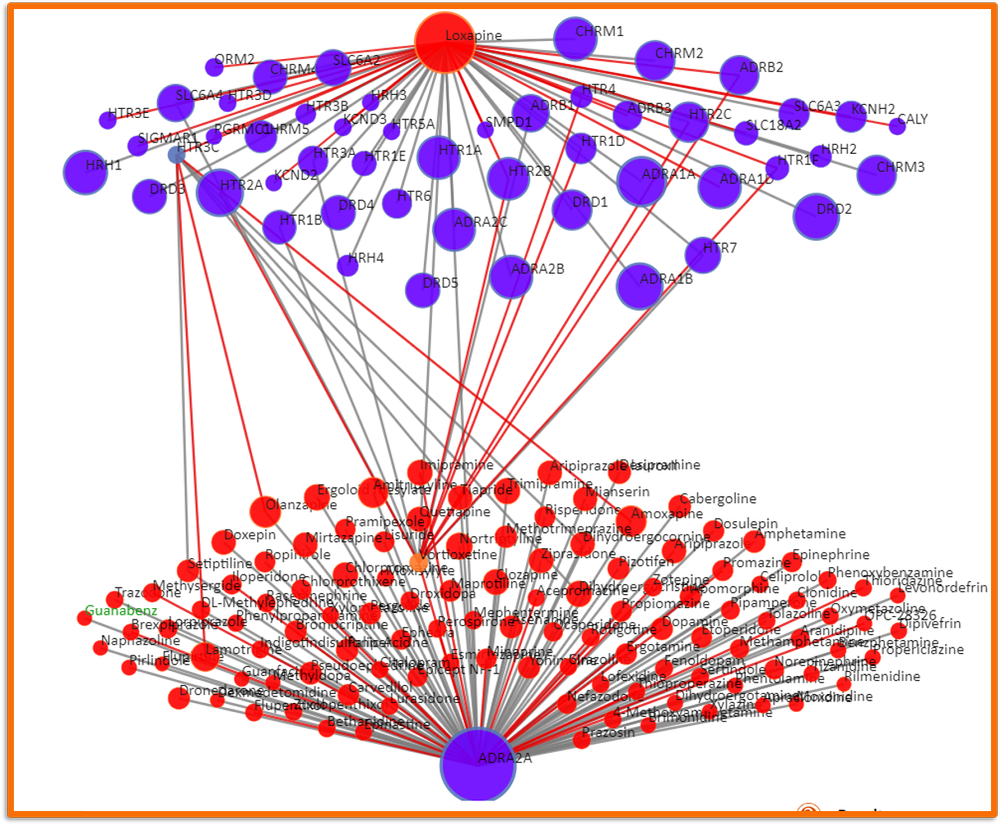
QuartataWeb-2: A Quantitative Systems Pharmacology (QSP) Platform for Drug–Target–Pathway–Disease Mapping
QuartataWeb-2 is the next generation of our platform, extending beyond drug–target interactions to provide a comprehensive systems biology resource. By integrating information from publicly available databases, it connects drugs, targets, pathways, and diseases into a unified framework. This expansion enables multi-scale exploration of therapeutic mechanisms, polypharmacology, and disease associations, offering a broader view of how molecular interventions propagate through biological networks.
At its foundation, QuartataWeb-2 employs advanced matrix factorization techniques and deep graph neural networks to infer novel associations and uncover hidden relationships within complex biological systems. These methods combine predictive accuracy with mechanistic interpretability, supporting drug discovery, repurposing, and the mapping of systems-level effects. QuartataWeb-2 thus positions itself as a versatile platform bridging pharmacology, network biology, and translational research.
Ref: Li H, Pei F, Taylor DL, Bahar I. (2020) QuartataWeb: Integrated Chemical-protein-pathway Mapping for Polypharmacology and Chemogenomics. Bioinformatics 36:3935-3937.
Simmerling, Kozakov, and Coutsias labs created hybrid approaches that combine physics-based sampling with AlphaFold model building and refinement
Traditional machine learning methods such as clustering have been a valuable part of the Laufer Center's toolkit for many years. With increases in computing power, and especially CPUs, we have now begun to incorporate deep learning approaches into our research. The Simmerling lab’s ff99SB protein force field is a component of the Nobel-prize winning AlphaFold model; more recently, Profs Kozakov, Coutsias and Simmerling have collaborated in the development of fine-tuned AlphaFold models for specialized systems. These methods are succeeding when AlphaFold alone fails, underscoring the role of physics where the experimental training data are too sparse to provide good predictions. We are also developing neural network approaches to improve the quality of our force fields and solvent models, enabled by our creation of large libraries of synthetic data for robust training and validation.
MHC-Fine: Fine-tuned AlphaFold for precise MHC-peptide complex prediction
Ernest Glukhov1 , Dmytro Kalitin1, Darya Stepanenko1, Yimin Zhu, Thu Nguyen, George Jones1, Taras Patsahan, Carlos Simmerling, Julie C. Mitchell, Sandor Vajda, Ken A. Dill, Dzmitry Padhorny, Dima Kozakov
Abstract: The precise prediction of major histocompatibility complex (MHC)-peptide complex structures is pivotal for understanding cellular immune responses and advancing vaccine design. In this study, we enhanced AlphaFold’s capabilities by fine-tuning it with a specialized dataset consisting of exclusively high-resolution class I MHC-peptide crystal structures. This tailored approach aimed to address the generalist nature of AlphaFold’s original training, which, while broad-ranging, lacked the granularity necessary for the high-precision demands of class I MHC-peptide interaction prediction. A comparative analysis was conducted against the homology-modeling-based method Pandora as well as the AlphaFold multimer model. Our results demonstrate that our fine-tuned model outperforms others in terms of root-mean-square deviation (median value for Cα atoms for peptides is 0.66 Å) and also provides enhanced predicted local distance difference test scores, offering a more reliable assessment of the predicted structures. These advances have substantial implications for computational immunology, potentially accelerating the development of novel therapeutics and vaccines by providing a more precise computational lens through which to view MHC-peptide interactions.
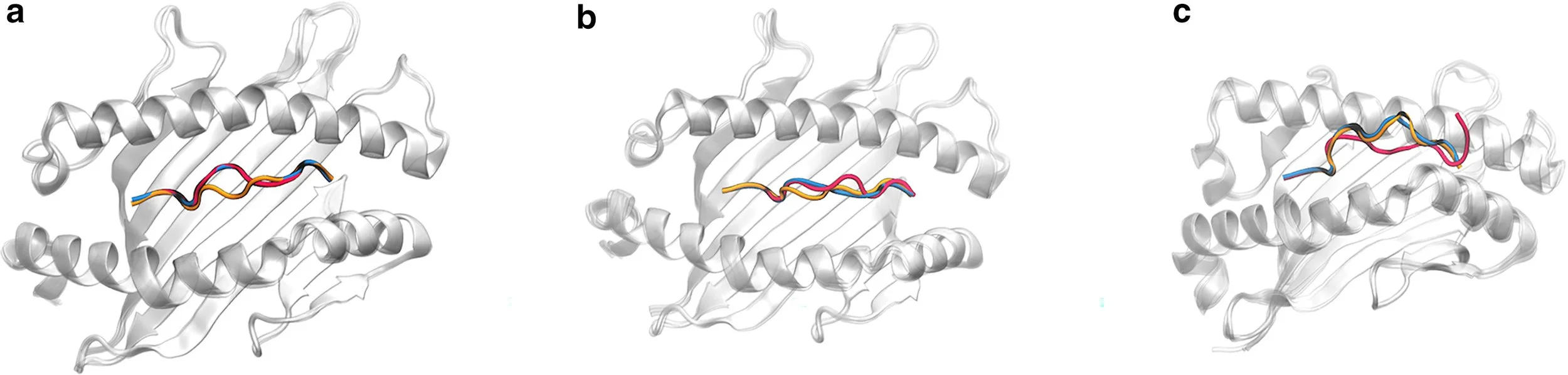
Comparative Visualization of pMHC Complex Prediction Accuracy.
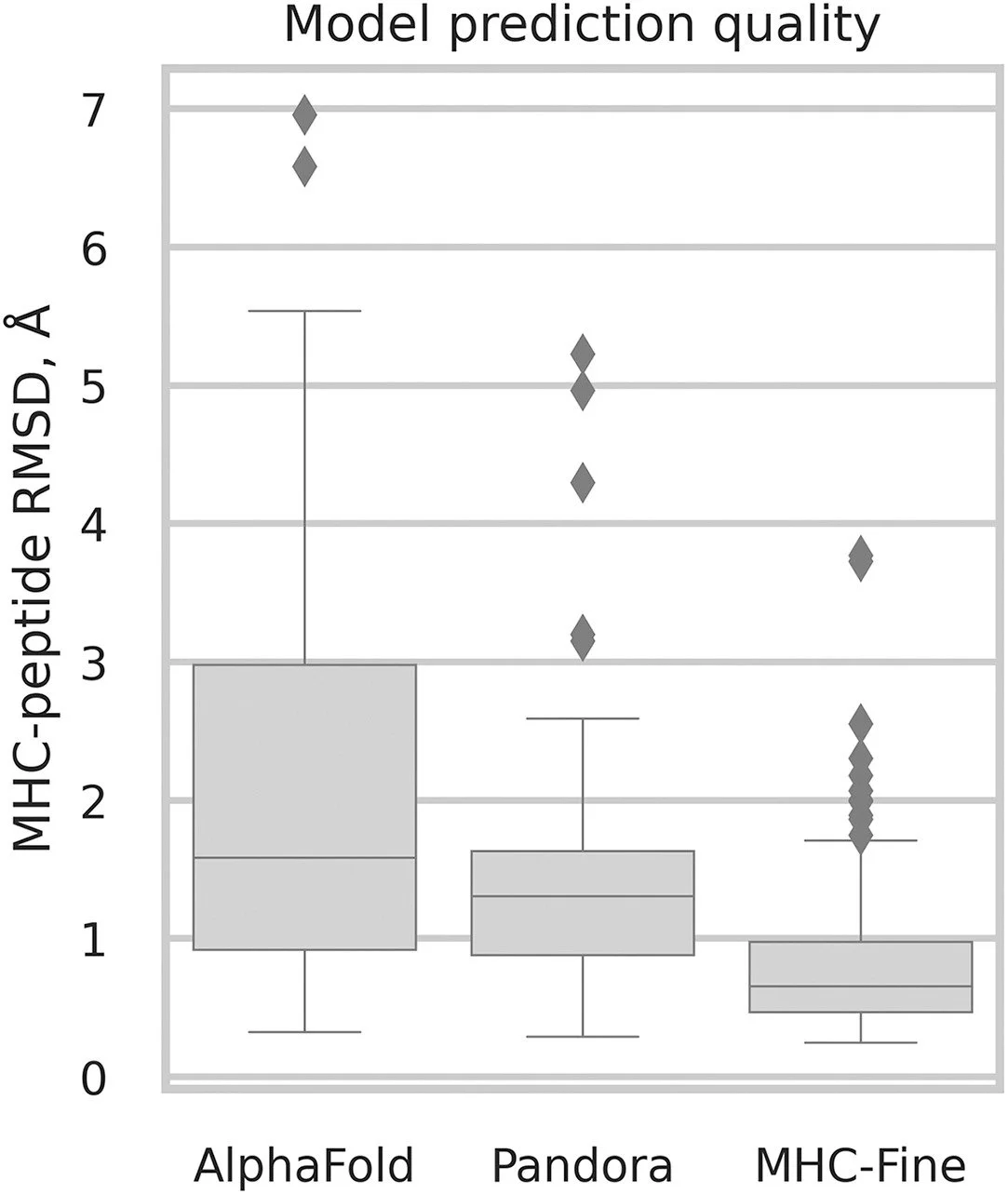
Comparative analysis of prediction accuracy for class I MHC-peptide complexes using RMSD calculated for Cα atoms of peptides: performance of AlphaFold, Pandora, and MHC-Fine.
Ref: MHC-Fine: Fine-tuned AlphaFold for precise MHC-peptide complex prediction. Glukhov et al. (2024) Biophys J. 123:2902-09.
All fields of science depend on mathematical models. Occam’s razor refers to the principle that good models should exclude parameters beyond those minimally required to describe the systems they represent. Mujica-Parodi lab showed how deep learning can be powerfully leveraged to apply Occam’s razor to model parameters.
Fixfit: A new Method for learning multi-scale brain dynamics features from functional neuroimaging

Antal BB, Chesebro AG, Strey HH, Mujica-Parodi LR, Weistuch C. Achieving Occam's razor: Deep learning for optimal model reduction. PLoS Comput Biol. 2024;20(7):e1012283. doi:10.1371/journal.pcbi.1012283.
Ref: Achieving Occam's razor: Deep learning for optimal model reduction
Antal BB, Chesebro AG, Strey HH, Mujica-Parodi LR, Weistuch C. Achieving Occam's razor: Deep learning for optimal model reduction. PLoS Comput Biol. 2024;20(7):e1012283. doi:10.1371/journal.pcbi.1012283.

Ref: Scientific Machine Learning of Chaotic Systems Discovers Governing Equations for Neural Populations
Chesebro AG, Hofmann D, Dixit V, Miller EK, Granger RH, Edelman A, Rackauckas CV, Mujica-Parodi LR, Strey HH. SarXiv:2507.03631 doi:10.48550/arXiv.2507.03631 (under review)